Perhaps one of the most fascinating classes of transporters, multidrug transporters function in the removal of structurally and chemically dissimilar cytotoxic molecules. Our targets belong to two transporter families ubiquitous in all three kingdoms of life. With a conserved function in efflux of hydrophobic cations, multidrug and toxic compound extrusion (MATE) antiporters have garnered intense interest in the last few years following a flurry of crystal structures. Human MATE1 and 2 carry out the final step in the excretion of metabolic waste and organic cations in the liver and kidney. The finding that MepA from S. Aureus, confers resistance against a new generation of glycylcycline antibiotics with promising activity towards methicillin- and vancomycin-resistant strains emphasized MATE transporters as important drug targets. The major facilitator superfamily (MFS) represents the largest evolutionarily-related collection of secondary active transporters in prokaryotes and eukaryotes. Cells expressing MFS-MDR transporters acquire resistance against a diverse group of unrelated toxic compounds leading to MDR phenotypes that hamper treatment of bacterial and fungal infections.
MATEs and MFS-MDR transporters catalyze obligatory ion/substrate antiport. The core tenants of the classic model of antiport are: 1) alternating access of the transporter between an IF conformation poised to bind substrates and an OF conformation that enables ion binding and 2) competition between ion and substrate. Competition can be direct, involving the mutually exclusive binding of ions and substrates to the same site, or allosteric wherein the two ligands bind at different locations but reduce each other’s affinity .
Our contribution
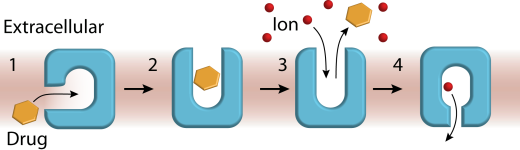
One of the longest ongoing in the lab, this research program encompassed investigations of representatives of small multidrug resistance (SMR), MFS and MATE transporter families and led to important conceptual contributions. We advanced novel transport models wherein substrate binding to a membrane-accessible conformation initiates the transport cycle by opening the extracellular side. Subsequent protonation of membrane-embedded acidic residues induces substrate release to the extracellular side and triggers conformational changes that conclude by proton release to the intracellular side (Scheme 1).
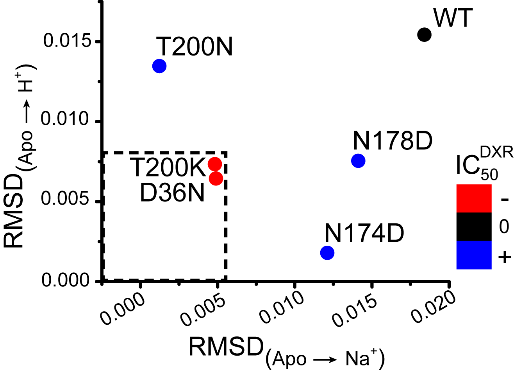
Our work on MATE transporters, NorM-Vc (V. cholera) and Pf-MATE (P. furiosus) as representatives of NorM and DinF subfamilies respectively, uncovered an interplay between Na+ and H+ in triggering conformational changes. We demonstrated that whereas Na+ allosterically modulates substrate affinity, H+ directly displaces the substrate from the binding site of NorM-Vc. Binding of the two ions to a site in the N-terminal lobe engenders opposite conformational changes of TM1 to which the ions are coordinated. The group of Faraldo-Gomez complemented our findings by computationally demonstrating that an equivalent cluster of residues in Pf-MATE is capable of binding Na+ and H+. Direct correlation between NorM-Vc ion-dependent structural dynamics with drug resistance activity illustrates how individual conserved residues modulate the transport energy landscape as illustrated below.