Current Projects
-
High-Resolution Ion Mobility Based on Traveling Wave Structures for Lossless Ion Manipulation Resolves Hidden Lipid Features

Investigator: Allison R. Reardon Lipids are an integral part of life with many functions spanning from energy storage and cell structure to essential hormones. Lipids have numerous isomers due to their acyl tail variability, which challenges analytical methods. As isomer systems become more complex, higher structural resolution is needed. Ion mobility coupled with mass spectrometry (IM-MS) is an effective technique for isomer separation, but drift tube IM (DTIM-MS), which serves as the IM “gold standard” performs moderate resolving power (Rp ~50) separations. High resolution IM (HRIM) based on traveling wave structures for lossless ion manipulation (TWSLIM) can access high resolving powers (Rp ~250) and can better address lipid isomer complexity.
To benchmark the performance of HRIM, we investigated several complex lipid isomer standards via TWSLIM-MS. These include geometric cis/trans isomers, double bond positional isomers, and geometric sn-position stereoisomers. In addition, more complex lipid samples consisting of total lipid extracts representing a variety of subclasses across the glycerophospholipid and sphingolipid classes were analyzed for their lipid isomer/isobar separation via DTIM-MS and TWSLIM-MS.
Our results indicate that some lipid features that were previously observed as a single IM peak with DTIM-MS exhibit additional IM peak features with HRIM-MS, suggesting these signals represent a mixture of isomers. Upon observing these enhanced lipid separations with HRIM-MS, the next challenge is to further identify the molecular identities of these newly-resolved features. The tentative lipid identifications are assigned using a combination of accurate mass, collision cross section (CCS), and lipid ion mobility-mass correlations, the latter of which utilizes empirically-derived structural trends mapping chain length and degrees of unsaturation.
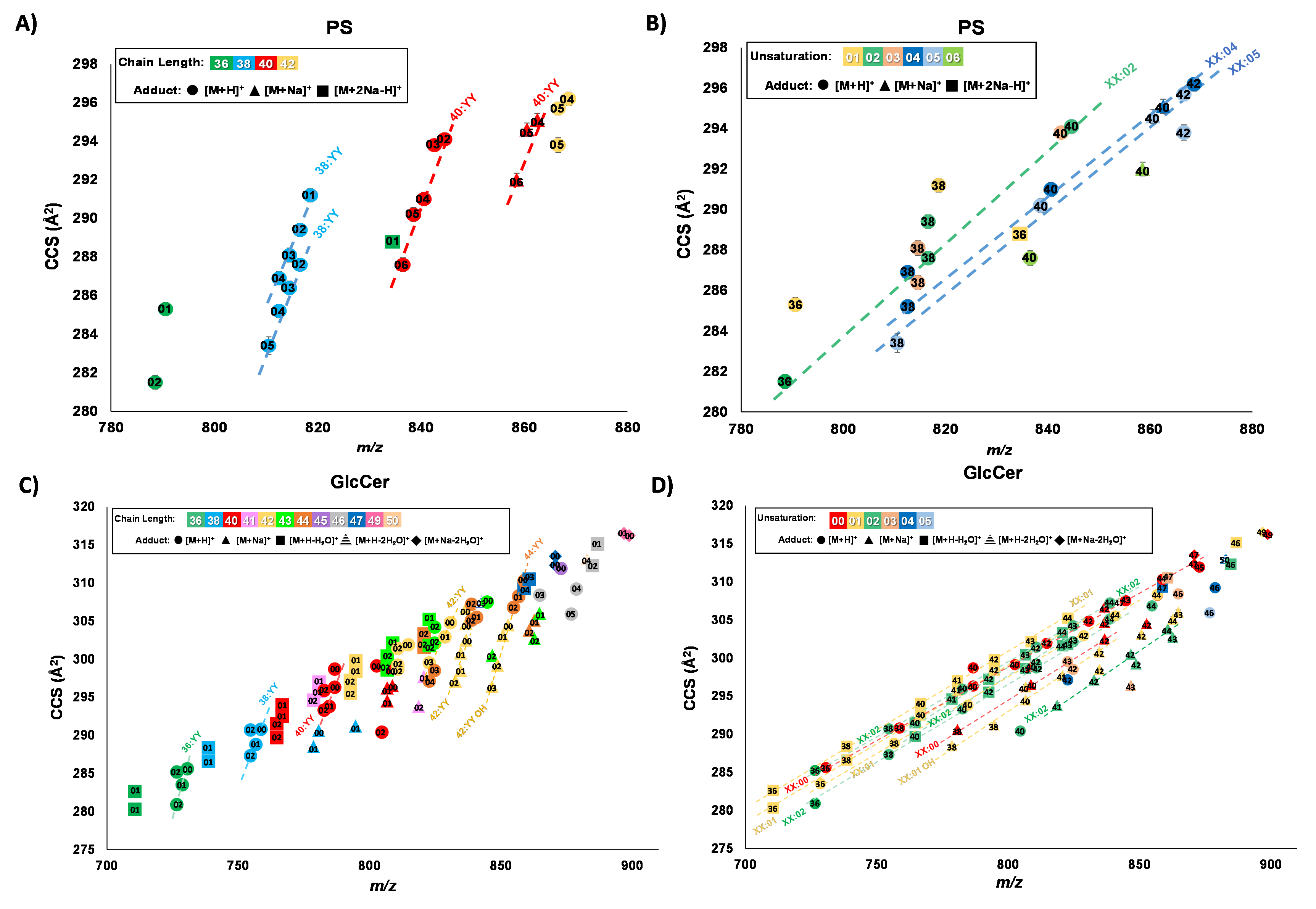
Figure: Mobility-mass correlations of PS and GlcCer total lipid extracts. A PS chain length linear correlations. B PS unsaturation linear correlations. C GlcCer chain length linear correlations. D GlcCer unsaturation linear correlations. A, C Chain length trendline identities labeled by total carbons on acyl tails:YY where YY indicates the total number of double bonds, with the number of double bonds annotated with the data points. B, D Unsaturation trendline identities are labeled by XX:total number of double bonds on the acyl tails where XX indicates the total number, with the number of carbons annotated with the data points. Trendlines each contain n ≥ 3 number of lipid features. Adduct type is designated by the shape of the marker.
Corresponding Publication
Reardon, A. R.; May, J. C.; Leaptrot, K. L.; & McLean, J. A. High-Resolution Ion Mobility Based on Traveling Wave Structures for Lossless Ion Manipulation Resolves Hidden Lipid Features. Anal. Bioanal. Chem. 2024.
-
Chiral Ion Mobility: Noncovalent Complexation Strategies to Enable Drug Enantiomer Differentiation using Ion Mobility-Mass Spectrometry (IM-MS)

Investigator: Benjamin K. Blakley There is considerable pharmaceutical and analytical interest in developing rapid methods which differentiate small molecule drug enantiomers. To this end, our group is working to develop structurally selective ion mobility-mass spectrometry workflows enabling the rapid differentiation of chiral drugs. Our workflows leverage high resolution ion mobility (HRIM) technologies to elucidate enantiomer-specific structural differences imparted during noncovalent complexation.
Previously, we demonstrated proof-of-concept for a chiral ion mobility workflow utilizing a ternary copper complex of the form [(M)(L-His)(CuII) – H]+ to differentiate chiral drugs and quantify enantiomeric excess. However, copper-histidine complexation did not result in the complexation and separation of all chiral drugs investigated. For this reason, more recently we have become interested in developing a broad-scale approach to chiral drug analysis which covers a broader range of drug classes and structures.
Currently, we are characterizing the separation capacity of a broad panel of chiral selectors, including metal-amino acid clusters, cyclodextrin inclusion complexes, and self-association multimers. This work, aided by collaborators at Pfizer, seeks ultimately to provide recommendations on where to start when assessing a given chiral drug using IM-MS. This project, while complex, has immense pharmaceutical applications, including drug development, quality control, and beyond.
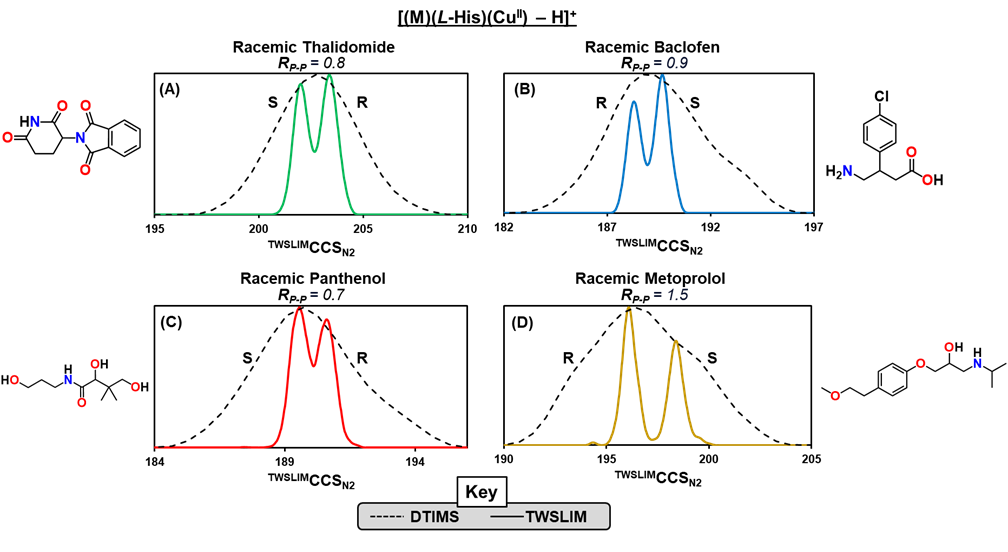
Figure: Combining noncovalent complexation with structurally selective HRIM technologies represents a powerful development in the field of enantiomer separations. HRIM analysis enables elucidation of four drug enantiomers coordinated within a ternary copper complex of the form [(M)(L-His)(CuII) – H]+. Separation is shown for racemic samples of (A) thalidomide, (B) baclofen, (C) panthenol, and (D) metoprolol. Notably, baseline separation was achieved for metoprolol.Corresponding Publication
Blakley, B.K.; Zlibut, E.; Gupta, R.M.; May, J.C.; McLean, J.A. Direct Enantiomer Differentiation of Drugs and Drug-Like Compounds via Noncovalent Copper-Amino Acid Complexation and Ion Mobility-Mass Spectrometry. 2024, Analytical Chemistry, 96(31), pp. 12892-12900.
-
Ion Mobility Resolution of Challenging Isomer Systems using High Resolution Demultiplexing: HRdm

Investigator: Jody C. May, Ph.D. Ion mobility separates ions based upon their effective size in the gas phase, and thus can differentiate isomers which have identical masses and thus cannot be separated by conventional mass spectrometry alone. For challenging isomer systems in which size differences are small, conventional ion mobility is oftentimes not enough to resolve isomeric peaks.
To address this, a new approach based upon a combined ion multiplexing and post-acquisition peak deconvolution strategy has been developed in collaboration with Agilent Technologies. Referred to as HRdm, this approach can achieve ion mobility resolving powers of greater than 200 at the highest processing level and has been demonstrated to resolve challenging isomeric systems. Currently this strategy is being optimized and applied to various isomeric systems in order to establish the application space for this technique.
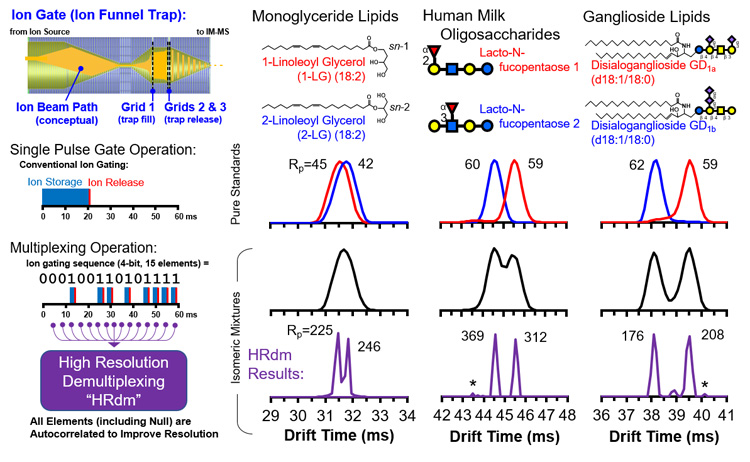 Figure: Ion multiplexing is implemented at the ion funnel trap on a commercial IM-MS instrument (6560, Agilent, upper left), where multiple ion pulses are introduced for ion mobility separation during each measurement cycle. Information from these multiple ion pulses are then autocorrelated (lower left) in order to determine signal and noise in the resulting IM spectrum. This HRdm approach yields higher ion mobility resolving power (Rp) than what can conventionally be achieved from the instrument, enabling isomeric mixtures to be resolved (right panels).
Figure: Ion multiplexing is implemented at the ion funnel trap on a commercial IM-MS instrument (6560, Agilent, upper left), where multiple ion pulses are introduced for ion mobility separation during each measurement cycle. Information from these multiple ion pulses are then autocorrelated (lower left) in order to determine signal and noise in the resulting IM spectrum. This HRdm approach yields higher ion mobility resolving power (Rp) than what can conventionally be achieved from the instrument, enabling isomeric mixtures to be resolved (right panels).Corresponding Publication
May, J.C., Knochenmuss, R., Fjeldsted, J.C. and McLean, J.A., 2020. Resolution of isomeric mixtures in ion mobility using a combined demultiplexing and peak deconvolution technique. Analytical Chemistry, 92(14), pp.9482-9492.
-
Effects of Solvent and Source Temperature on Ion Mobility Collision Cross Section Measurements

Investigator: Nadjali A. Chung Ion mobility (IM) is a gas-phase electrophoretic technique that separates ions on the basis of size, shape, and charge, reflected in the IM-derived collision cross section (CCS) value. CCS values represent the rotationally averaged surface area of ions and were found to be reproducible and robust when compared across different low-field drift tube IM (DTIMS)-MS systems.
Recent works have taken advantage of the high reproducibility of CCS values to compile libraries in support of compound identification. To effectively use these CCS libraries, IM parameters need to be standardized, accounting for sources of measurement variation. While many drift tube parameters were assessed in previous work, the effects of solvent and ion source temperatures have not been rigorously documented for small molecules. To address this, this work assessed changes in CCS in different molecular classes across discrete changes in solvent composition and source temperatures.
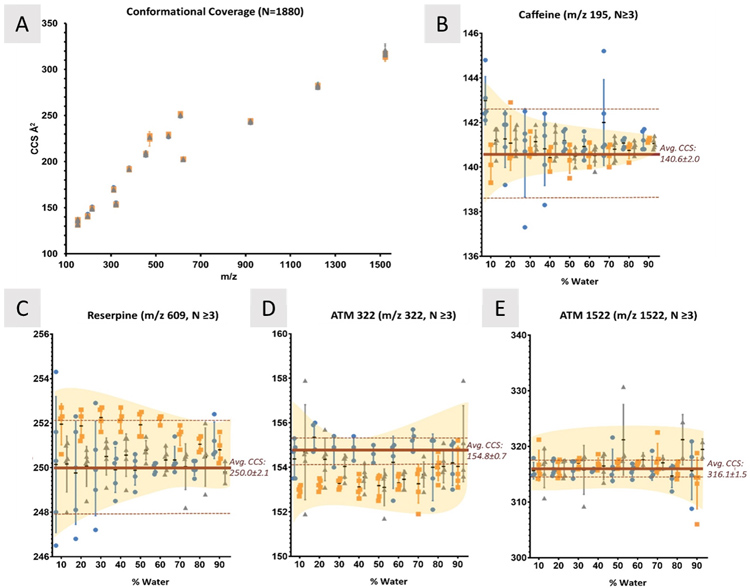
Figure 1. Changes in CCS between solvent composition. A) CCS measurements for 15 compounds (m/z 152-1522) were measured in 27 different solvent systems with 4 or 5 replicates each. As the amount of water in each system is increased, CCS values also change, but not in a consistent manner between compounds. In the analysis of caffeine (B) the solvent systems at 10% water have significantly different CCS values, but as the water content increases, each converge to about 141 Å2. Compared to reserpine (C), however, there is no significant convergence to a single value between any of the solvent systems, similar to ATM 322 (D). E) ATM 1522 shows little deviation from the accepted CCS regardless of solvent system, which supports the hypothesis that molecular interactions with each of the solvents also plays an important role in any solvent effects observed. IPA systems contain N=4. Yellow shaded regions highlight the general trend of changes observed. Significance was determined using Kruskal-Wallis analyses; data was unmatched, assumed nonparametric, and the mean rank of each solvent was compared to the other. Dunn’s correction for multiple comparisons was applied, and α=0.05.
Corresponding Publication
Chung, N.A., May, J.C., Robinson, R.A. and McLean, J.A., 2023. Solvent Composition Can Have a Measurable Influence on the Ion Mobility-Derived Collision Cross Section of Small Molecules. Journal of the American Society for Mass Spectrometry, 35(2), pp.234-243.