Research
How does nucleotide biosynthesis regulate adipogenesis?
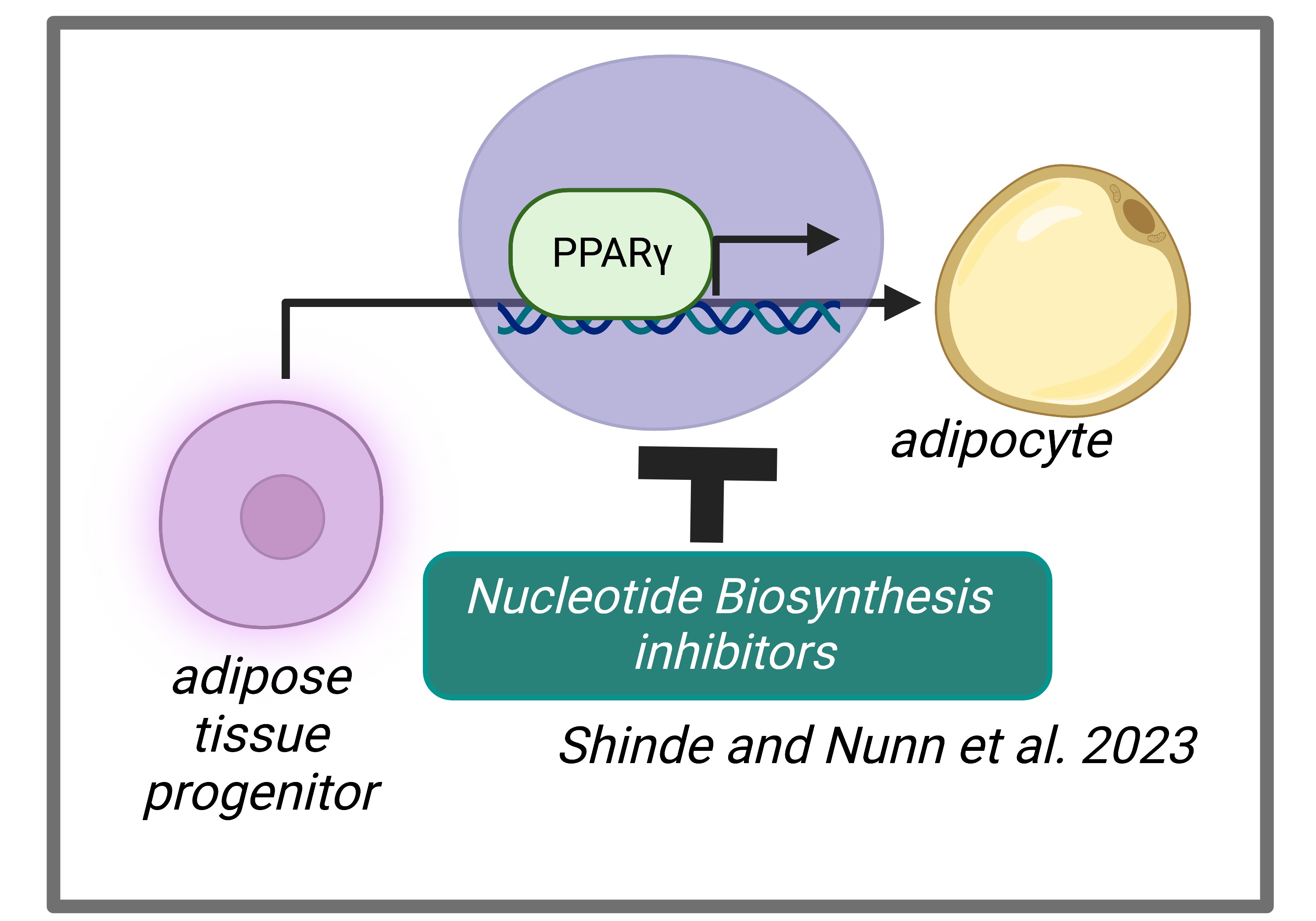
Energy balance and nutrient availability are key determinants of cellular decisions to remain quiescent, proliferate or differentiate into a mature cell. After assessing its environmental state, the cell must rewire its metabolism to support distinct cellular outcomes. Mechanistically, how metabolites regulate cell fate decisions is poorly understood. We used adipogenesis as our model system to ascertain the role of metabolism in differentiation. We isolated adipose tissue stromal vascular fraction (SVF) cells and profiled metabolites before and after adipogenic differentiation to identify metabolic signatures associated with these distinct cellular states. We found that differentiation alters nucleotide accumulation. Furthermore, inhibition of purine, and to a lesser degree pyrimidine, biosynthesis prevented lipid storage within adipocytes and downregulated the expression of lipogenic factors. In contrast to proliferating cells, in which mTORC1 is activated by purine accumulation, mTORC1 signaling was unaffected by purine levels in differentiating adipocytes. Rather, our data indicated that purines regulate transcriptional activators of adipogenesis, PPARγ and C/EBPα to promote differentiation. Although de novo purine biosynthesis has mainly been studied in proliferation, our study points to a requirement of this metabolic pathway in cell differentiation. (Shinde and Nunn et al., 2023).
Our new projects of interest are to:
- Determine the fate of cells that fail to differentiate into adipocytes when exposed to inhibitors of nucleotide biosynthesis.
- Elucidate the underlying mechanism by which blocking nucleotide biosynthesis disrupts the function of PPARγ.
- Evaluate the impact of inhibitors of nucleotide biosynthesis on adipogenesis in vivo and assess the consequent effects on metabolic health.
By examining these aspects, we aim to gain a comprehensive understanding of the relationship between nucleotide biosynthesis, PPARγ function, adipogenesis, and metabolic health.
How does mitochondrial metabolism drive cellular decisions?
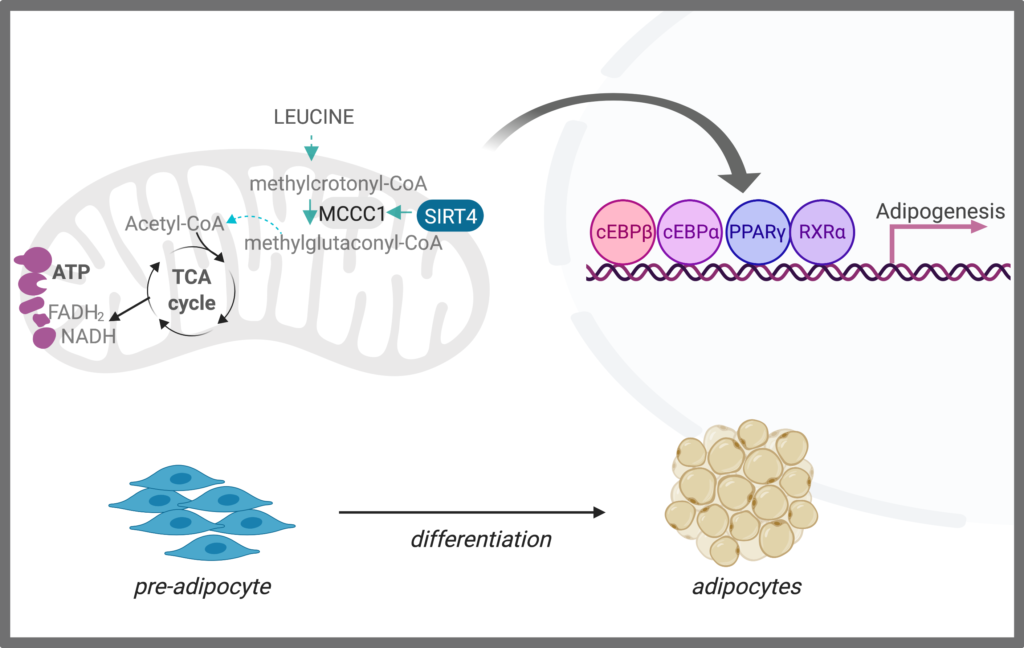
We aim to understand fundamental questions on adipose biology that are still unanswered: how do mitochondria modulate adipogenesis? How can metabolism be leveraged to induce healthy adipose expansion that allows for excess nutrient storage, preventing insulin resistance under a stress of a high fat diet? We found that branched-chain amino acid (BCAA) catabolism is upregulated early in the differentiation process, and this regulation is not dependent on transcriptional activity of a key adipogenic regulator, transcription factor PPARγ. Rather, a mitochondrial sirtuin, SIRT4 promotes BCAA catabolism to induce a nuclear transcriptional program that drives adipogenesis (Zaganjor et al., 2021).
Our new projects of interest are to:
- Examine how mitochondria communicate with the nucleus to promote adipogenesis.
- Define new mechanisms by which mitochondria activate early adipogenesis using CRISPR screening.
- Evaluate the impact of altering mitochondrial metabolism on non-cell autonomous processes using in vivo transgenic and knockout mouse models.
Mitochondrial function in cancer
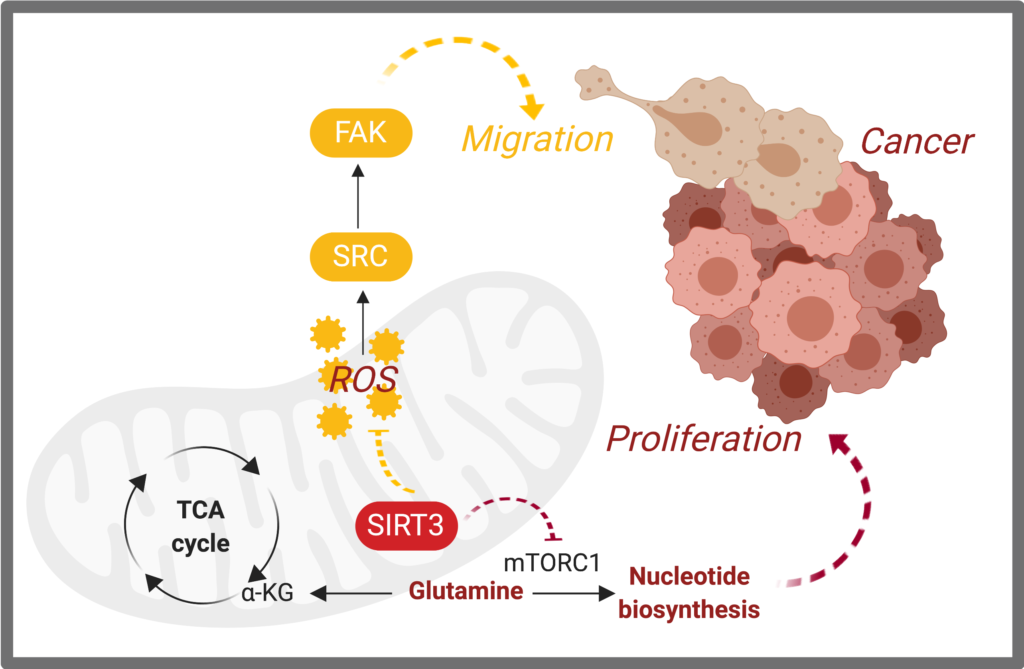
Metabolic rewiring that supports tumor initiation, progression, metastasis, resistance to chemotherapy, and even immune evasion has been at the center of cancer biology research. Along these lines, we identified critical mitochondrial signaling events that promote cancer growth and metastasis. We found that a sirtuin, SIRT3, a global regulator of mitochondrial metabolism, is down-regulated in basal-like breast cancers, causing these cancers to rewire glutamine metabolism for nucleotide synthesis, driving proliferation (Gonzalez Herrera*, Zaganjor*, et al. 2018). Moreover, we discovered that SIRT3 regulation of reactive oxygen species blocks breast cancer metastasis through Src signaling (Lee*, van de Ven*, Zaganjor*, et al. 2018).
Our new projects aim to:
- Elucidate the direct mechanism by which SIRT3 promotes nucleotide synthesis.
- Develop screening tools to identify the mechanisms by which loss of SIRT3 promotes migration and metastasis.
- Evaluate new strategies to target low-SIRT3 cancer cell proliferation and migration.