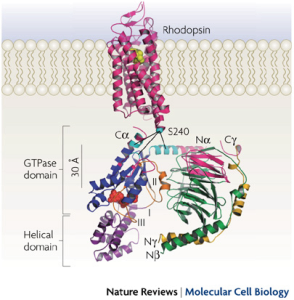
 My work is focused on understanding the molecular basis of signaling mechanisms mediated by G proteins, which are switch proteins. G proteins are normally inactive, but a receptor that has received a specific signal can activate G proteins, leading to changes in the activity of enzymes that produce second messengers such as cyclic AMP and calcium. The resulting changes in cellular activity underlie a large number of physiological processes. G protein-mediated signalling cascades are key regulators of many physiological processes, including processes of development, differentiation, and regulation of cell division. In the brain, many key neurotransmitters and neuromodulators mediate a myriad of functions by activation of such G protein cascades. The research in my laboratory is aimed at understanding how G proteins become activated by receptors, how they in turn activate effector enzymes, and how they turn off. We determined the sites of interaction between proteins using a method of decomposing the proteins into small synthetic peptides and determining which peptides blocked interaction sites (Hamm et al., 1988; Rarick et al., 1992; Artemyev et al., 1993; Arshavsky et al., 1994). To understand the process more fully, we determined the atomic structure of the proteins in collaboration with the group of Paul Sigler. We used X-ray crystallography to solve the three-dimensional structures of G proteins in their inactive (GDP bound), (Lambright et al., 1994) and activated (GTPγS-bound) forms (Noel et al., 1993). We caught a glimpse of the self-inactivating process in another crystal form, the transition state analog, Gα.GDP.AlF4- (Sondek et al., 1994). Soon after, the structures of the βγ subunit (Sondek et al., 1996) and the heterotrimeric G protein (Lambright et al., 1996) were solved. These high-resolution structural studies allowed us to postulate specific hypotheses regarding mechanisms of receptor:G protein interaction and activation, G protein subunit association-dissociation and effector activation.
My work is focused on understanding the molecular basis of signaling mechanisms mediated by G proteins, which are switch proteins. G proteins are normally inactive, but a receptor that has received a specific signal can activate G proteins, leading to changes in the activity of enzymes that produce second messengers such as cyclic AMP and calcium. The resulting changes in cellular activity underlie a large number of physiological processes. G protein-mediated signalling cascades are key regulators of many physiological processes, including processes of development, differentiation, and regulation of cell division. In the brain, many key neurotransmitters and neuromodulators mediate a myriad of functions by activation of such G protein cascades. The research in my laboratory is aimed at understanding how G proteins become activated by receptors, how they in turn activate effector enzymes, and how they turn off. We determined the sites of interaction between proteins using a method of decomposing the proteins into small synthetic peptides and determining which peptides blocked interaction sites (Hamm et al., 1988; Rarick et al., 1992; Artemyev et al., 1993; Arshavsky et al., 1994). To understand the process more fully, we determined the atomic structure of the proteins in collaboration with the group of Paul Sigler. We used X-ray crystallography to solve the three-dimensional structures of G proteins in their inactive (GDP bound), (Lambright et al., 1994) and activated (GTPγS-bound) forms (Noel et al., 1993). We caught a glimpse of the self-inactivating process in another crystal form, the transition state analog, Gα.GDP.AlF4- (Sondek et al., 1994). Soon after, the structures of the βγ subunit (Sondek et al., 1996) and the heterotrimeric G protein (Lambright et al., 1996) were solved. These high-resolution structural studies allowed us to postulate specific hypotheses regarding mechanisms of receptor:G protein interaction and activation, G protein subunit association-dissociation and effector activation.
There are a number of projects ongoing in the laboratory; rotation projects as well as postdoc positions are available in several areas:
1. Protease activated receptor function in the cardiovascular system and other diseases
2. Regulation of synaptic transmission by Gβγ-mediated inhibition of exocytotic fusion
Protease Activated Receptor 4 (PAR 4)
Thrombin is the major protease in the coagulation cascade whose pleiotropic actions can ultimately lead to thrombosis and tissue injury. Thrombin works by activation of the G protein-coupled protease activated receptors PAR1 and PAR4 on human platelets to initiate signaling cascades leading to increases in [Ca2+i], secretion of autocrine activators, trafficking of adhesion molecules to the plasma membrane, “inside-out” integrin activation and shape change, which all promote platelet aggregation. We have uncovered a number of differences in signaling properties of PAR1 and PAR4 receptors in platelets (M. Duvernay, Young, Gailani, Schoenecker, & Hamm, 2013; M. T. Duvernay, Temple, Maeng, Blobaum, Stauffer, Lindsley, & Hamm, 2017). The thrombin receptors work in a progressive manner, with PAR1 activated at low thrombin concentrations, and PAR4 recruited at higher thrombin concentrations implying distinct roles for the two receptors in thrombosis and hemostasis. Indeed, a complete pharmacological profiling of PARs on platelets revealed superior efficacy downstream of PAR4 in comparison to PAR1. PAR4 being engaged after PAR1 and displaying superior efficacy suggest it may be a better pharmacological target that may reduce the thrombotic burden while leaving hemostasis intact. However, the lack of an efficient PAR4 antagonist has prevented an accurate assessment of PAR4’s role, therefore we initiated efforts to target PAR4 with a small molecule.
Drug Discovery: PAR4 Antagonist Development
PAR4 is a promising drug development target for anti-thrombotic pharmaceuticals. Compounds developed by Bristol-Myers-Squibb (BMS) demonstrated the proof of concept that inhibition of PAR4 is an effective strategy for thrombosis and notably PAR4 antagonism carried much lower liability for bleeding than all other established anti-platelet drugs. However the BMS compounds have poor bioavailability and better PAR4 antagonists are need both for clinical development and for tool compounds to study PAR4 biology in animal models of diseases.
We generated structure activity relationships (SAR) around a novel PAR4 inhibitor chemotype identified from a small molecule library curated by the Vanderbilt High Throughput Screening Core (Bertron et al., 2021). The IC50 values against PAR4 agonist peptide (PAR4-AP) in human platelets have been driven from 3.4 µM from the original hit down to 66 nM. There is measurable free fraction in our series (0.02), in contrast to the BMS PAR4 inhibitor which was undetectable in the free fraction in our hands. While many compounds in our compound families were highly effective against the synthetic peptide agonist PAR4-AP, antagonist activity against the tethered ligand generated by γ-thrombin cleavage of PAR4 remained a challenge.
As our earlier generations of compounds were highly effective against the PAR4 agonist peptide but not the thrombin-generated tethered ligand, we collaborated with Dr. Jens Meiler to leverage computational modeling and ultra-large library virtual screening to specifically target the tethered ligand focusing on compounds predicted to bind deeper in the agonist binding pocket. We have adopted an approach driven by in silico ultra-large library screening of a compound library available for make-on-demand parallel synthesis from a commercial source. Preliminary data demonstrate the feasibility of this approach for PAR4 searching a virtual space of up to 21,000,000,000 compounds. A pilot biological screen of 80 synthesized compounds identified a validated lead compound with selectivity for inhibition of thrombin activation of PAR4 over PAR1. We have thus far improved potency of this lead from γ-thrombin inhibition IC50 2.3 µM to an analog with an IC50 of 130 nM. While existing PAR4 chemotypes from our own project as well as other sources (including the BMS compounds that entered early clinical trials) broadly display strong potency preference for inhibition of human PAR4 over mouse PAR4, our current best compound has an IC50 of 255 nM for γ-thrombin PAR4 activation in mouse platelets, providing evidence that our approach is making progress on development of an inhibitor of both human PAR4 and mouse PAR4. Compounds meeting benchmark properties for potency, selectivity, and DMPK will be advanced to in vivo investigation.
Cerebrovascular involvement in Alzheimer’s Disease: PAR4 Antagonism
Because traumatic brain injury is a major risk factor for AD, wounding-induced platelet activation and thrombin are at the top of a chain of events leading to fibrin deposition, microinfarcts, blood-brain barrier disruption and inflammation that may contribute to vascular cognitive impairment and dementia (VCID). PAR4 contributes most of the platelet-derived thrombin, greater procoagulant microparticle formation, increased fibrin deposition, and initiation of platelet- stimulated inflammation. PAR4 is also expressed in immune cells and vasculature, and under inflammatory conditions, PAR4 is overexpressed via epigenetic demethylation of the PAR4 gene, F2RL3. PAR4 knockout studies have determined a role for PAR4 in hemostasis and thrombosis, as well as in neutrophil homing and invasion at the site of vascular insult. We have shown higher levels of F2RL3 expression in the prefrontal cortex in Alzheimer’s Disease dementia as compared to participants with normal cognition. Higher levels of F2RL3 expression in the prefrontal cortex of aging adults were associated with a faster rate of cognitive decline in a longitudinal human cohort evaluating cognitive aging, the Religious Orders Study (ROS) and the Rush Memory and Aging Project (MAP). Furthermore, F2RL3 expression correlates with expression of inflammatory markers in the prefrontal cortex. We also examined the role of PAR4 in the 5xFAD mouse AD model by comparing 5xFAD mice with WT PAR4 to 5xFAD mice with PAR4 KO. In the mouse model we find that 1) PAR4 expression is increased in the microvasculature of 5xFAD mice compared to WT mice and 2) Fibrin deposition is reduced in 5xFAD/PAR4KO compared to 5xFAD/WT mice. We are currently testing whether PAR4KO protects against the learning and memory deficits of the 5xFAD mice during aging. These studies will test whether PAR4 should be considered as a potential therapeutic target for vascular contributions to cognitive diseases.
Protease activated receptor 4 (PAR4) contributes to acute kidney injury
In addition to its role on platelets, PAR4 is expressed on other cell types including endothelial cells. Under inflammatory conditions, PAR4 is overexpressed via epigenetic demethylation of the PAR4 gene, F2RL3. While PAR4 has been reported to be expressed within the kidney, the contribution of PAR4 to acute kidney injury (AKI) and chronic kidney disease (CKD) is not well understood. We have found that PAR4 KO mice are protected against fibrosis following kidney injury in two mouse models. First, PAR4 KO mice are protected against induction of markers of both fibrosis and inflammation seven days following unilateral ureteral obstruction (UUO). We further show that PAR4 expression in the kidney is low in control mouse kidney but induced over time following UUO. Second, PAR4 KO mice are protected against kidney function pathologies in an AKI-CKD transition model of ischemia reperfusion followed by contralateral nephrectomy. While PAR4 is notable for the key role it plays in platelet activation and thrombosis, the role of PAR4 in other cell types and in the pathophysiology of a range of diseases is emerging. PAR4 signaling on platelets and/or cells intrinsic to the kidney may contribute to kidney pathology and pharmacological inhibition of PAR4 may be a therapeutic target for some types of kidney disease.
Regulation of synaptic transmission by Gβγ mediated inhibition of exocytotic fusion
We are investigating the molecular basis for interactions of Gαβγ subunits as well as βγ interaction with downstream effectors. It is known that a number of signaling pathways are regulated by free βγ subunits, which are liberated by the GTP-dependent dissociation from the α subunit. Some of these are K+ and Ca2+ channels, phospholipase Cβ, PI-3-kinase, certain isoforms of adenylyl cyclase, βARK, and MAP kinase cascades. Site-directed Ala scanning mutagenesis was used to characterize the molecular basis of these interactions (Ford et al., Sci 280, 1271, 1998). In addition to these, there are possibly other effectors of Gβγ subunits, and we are seeking to discover novel effectors using yeast two-hybrid techniques. We have recently shown that Gβγ subunits interact with SNARE proteins which make up the fusion machinery for exocytosis, a novel Gβγ effector (Blackmer et al., Nature Neurosci 8, 421, 2005; Yoon et al., Mol Pharmacol, 72, 1210, 2007).
One of the most important roles of Gβγ subunits in the brain is to monitor and control the amount of neurotransmitter release at synapses by Gi/o-coupled inhibitory neurotransmitter receptors. They do this through a dual regulation of the amount of Ca2+ coming in through voltage gated Ca2+ channels [Ford et al., Sci 280, 1271, 1998], and direct inhibition of synaptotagmin binding to SNARE proteins, the exocytotic fusion apparatus (Blackmer et al., Sci, 292, 293, 2001; Blackmer et al., Nature Neurosci 8, 421, 2005; Gerashchenko et al., Nature Neurosci., 8, 597, 2005] . Synaptotagmin is the Ca2+ sensor that triggers SNARE assembly and exocytosis. Gβγ directly competes with synaptotagmin binding to SNAP25 and syntaxin, two of the SNARE proteins [Yoon et al., Mol Pharmacol, 72, 1210, 2007]. This results in an inhibition of vesicular fusion, as well as a change in vesicle fusion mode to “kiss-and-run” [Photowala et al., Proc Natl Acad Sci USA, 103, 4281, 2006]. We believe that this is one of the regulatory mechanisms the neural and endocrine systems use to modulate hormone and neurotransmitter release; in the brain, this is a key mechanism for modulating neural circuitry in information processing. We are interested in this mechanistically, and we have worked out the details of how these proteins interact Wells et al., [Mol Pharmacol. 82,1136, 2012; Zurawski et al., Mol Pharmacol.89, 75, 2016]. Gβγ inhibits vesicular fusion even in a completely reconstituted system [Zurawski et al., 2017 J Biol Chem. 292, 12165].
Recently, we showed that removal of the C-terminal 3 amino acids from SNAP25 decreases Gβγ-SNARE interaction (Zurawski et al., Mol Pharmacol. 89, 75, 2016), and based on this finding, we have generated CRISPR mice with a premature stop codon. Mice defective in Gβγ-SNARE interaction allow us to examine the in vivo importance of this mechanism downstream of calcium entry. The mutant mice exhibit a number of electrophysiological and behavioral deficiencies, highlighting this interaction as a critical regulatory mechanism for hormone or neurotransmitter release phenotypes (Zurawski et al., bioArxiv, 2018 http://dx.doi.org/10.1101/280347 ). Importantly, this mouse model shows that the two main inhibitory presynaptic mechanisms (Gbg modulation of calcium entry, and Gbg inhibition of exocytosis through direct interaction with the SNARE complex) are distinct and separately addressable in vivo. Most interestingly, these two mechanisms are synergistic with each other, a completely unexpected result. This observation raises the possibility that therapeutic pairing of drugs that affect each mechanism may themselves work synergistically, an exciting possibility.
Also, we are preparing small molecule inhibitors and enhancers of the Gβγ-SNARE interaction to have an acute test of the importance of this regulatory step in synaptic physiology.
Biophysical and structural studies of G protein signaling

How does a G protein-coupled receptor interact with the heterotrimeric G protein? What are the contact sites, and how does it work at a distance to catalyze GTP/GDP exchange? What is the path through the molecule that ultimately causes GDP release? Dynamic measures of the conformational changes underlying signaling require a number of solution biophysical methods, such as fluorescence and electron paramagnetic resonance. We are using the approach of engineering in Cys residues into Cys-less mutant G proteins that can then be targeted with fluorescent groups or nitroxides (Yang et al., JBC, 274, 2379, 1999). Such engineered G protein molecules can be monitored constantly as they undergo conformational changes during the activation and inactivation process. Injection of fluorescently labeled G protein subunits into cells allows trafficking to specific sites, and dynamic changes in their localization. Double Cys mutants can be used in fluorescence resonance energy transfer as well as electron paramagnetic resonance experiments to provide information about changes in intra- or inter-molecular distances during signaling, and cross-linking studies can assess the relevance of the conformational changes for the signaling process. Upon GTP binding to the G protein α subunit, what drives dissociation from the receptor? These studies have been published in a number of papers, notably in Medkova et al., Biochem. 41, 9962, 2002; Preininger et al., Biochem. 42, 7931, 2003; Oldham et al., Nat Struct Mol Biol 13, 772, 2006; Oldham et al., Proc Natl Acad Sci USA 104, 7927, 2007; Van Eps et al., Proc Natl Acad Sci USA 103, 16194, 2007; Preininger et al., Biochem., 47, 10281, 2008; Preininger et al., Biochem., 48, 2630, 2009, Van Eps et al., Proc Natl Acad Sci USA 108,9420,2011; Alexander et al., Nat Struct Mol Biol. 21,56 2013; Kaya et al., J Biol Chem. 289,24475, 2014; Kaya et al., J. Biol. Chem. 2016; reviewed in Oldham and Hamm, Nat Rev Mol Cell Biol, 9, 60, 2008.
Mathematical modeling of G protein signaling networks
Various physiological responses appear to be controlled by multiple G proteins suggesting a network of interactions between multiple G protein pathways. Cells respond to a variety of signals, and cellular responses are really network responses that need network approaches to analyze them within the context of integrated physiological responses. Thus we have begun to identify systems functions of G protein pathways within the context of integrated physiological responses, and we are applying mathematical modeling approaches to understand these networks of G protein signaling pathways. We have been doing iterative experimentation and modeling studies on two main problems, visual transduction in rod photoreceptors (Andreucci et al., Biophys J 85, 1358, 2003; Caruso et al., Syst Biol, 152, 119, 2005; Caruso et al., Biophys J 91, 1192, 2006; Bisegna et al., Biophys J 94, 3363, 2008; Shen et al., IET Syst Biol 4, 12, 2010; Caruso et al., Proc Natl Acad Sci USA, 108, 7804, 2011) and the more complex signaling in endothelial cells (McLaughlin et al., J Biol Chem, 280, 25048, 2005; Lenoci et al., Mol Biosyst 7, 1129, 2011) .
The combined structural, functional and systems information will contribute to our understanding of basic mechanisms of cellular activation by a variety of signals, and will also provide insight into diseases that affect G protein-mediated function.