Arrestins in Cell Death
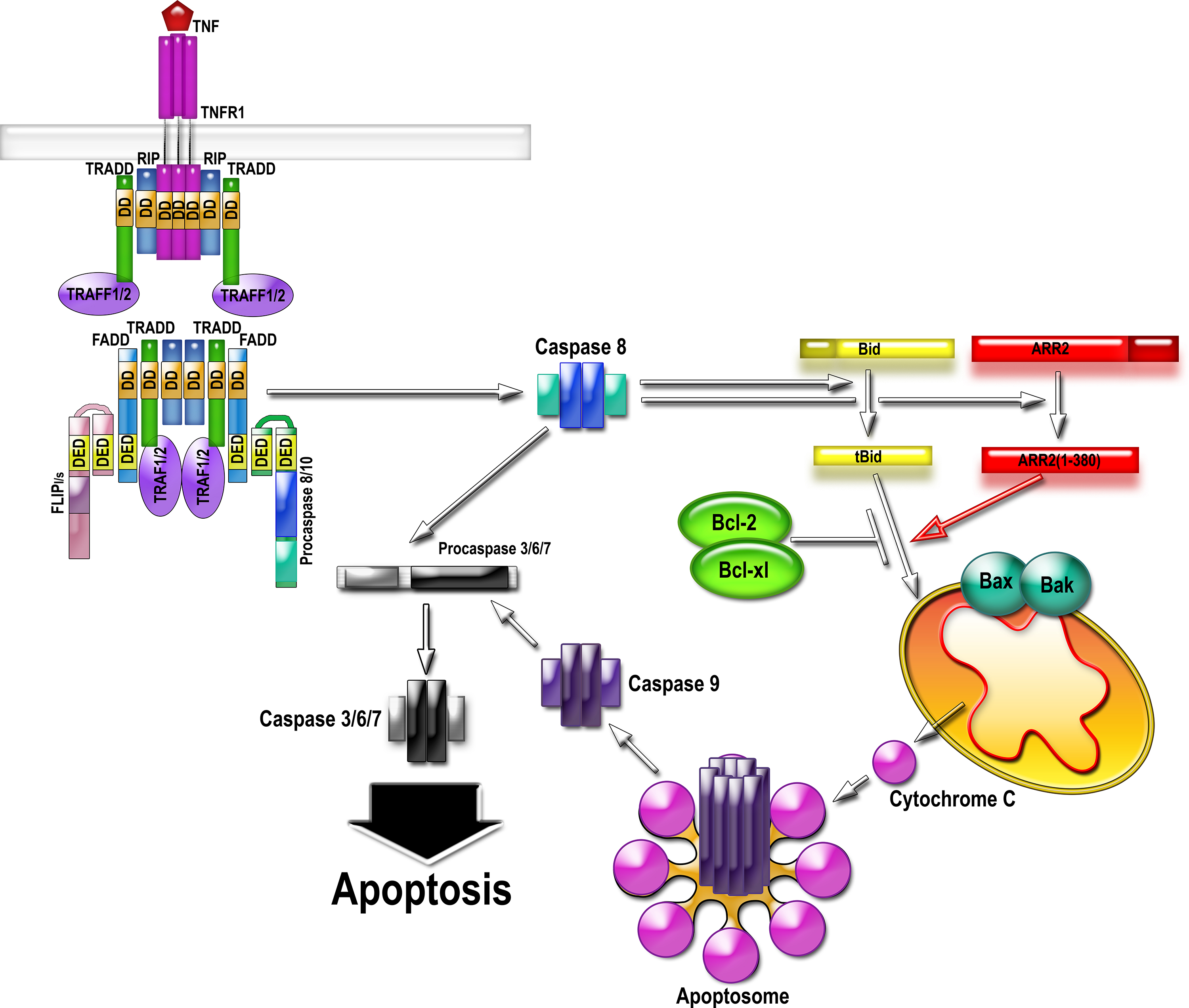
The activation of the death receptor by TNFalpha results in the assembly of the multi-protein complex II that directly activates caspase-8. Caspase-8 cleaves arrestin-2 and BID, generating 1-380 and tBID. 1-380 directly binds tBID and greatly enhances its ability to induce cytochrome C release from mitochondria. Cytochrome C organizes apoptosome, activating caspase-9, which then activates massive amounts of caspase-3. Therefore, cytochrome C release considered “the point of no return” in cell commitment to death is facilitated via interaction of tBID with caspase-generated arrestin-2 fragment 1-380. Abbreviations: TNFR, TNFalpha receptor; RIP, Receptor-interacting serine/threonine-protein kinase 1; FADD, Fas-associated death domain protein; TRADD, TNF receptor-associated death domain (TRADD); TRAF, TNF receptor-associated factor; FLIP, FLICE-like inhibitory protein (a.k.a. CFLAR, CASP8 and FADD-like apoptosis regulator); DD, death domain; DED, death effector domain.
Our lab is pursuing an exciting story about a direct role of arrestin2 in apoptotic cell death. Arrestin2 is cleaved by caspases during apoptosis initiated by multiple agents in many cell types. It seems that arrestin2 is preferentially cut by caspase-8, which is activated as part of a complex induced by the activation of death receptors in response to factors such as TNFalpha (tumore necrosis factor alpha). The resulting arrestin2 N-terminal cleavage fragment accumulates in the mitochondria and promotes, in cooperation with trancated BID, tBid (which, incidentally, is also cut by caspase-8), cytochrome C release, thereby propagating caspase activation and promoting cell death.
Interestingly, arrestin2 interacts with parkin, a protein implicated in Parkinson’s disease. Parkin was discovered in 1998 as the protein encoded by the gene mutated in families with inherited Autosomal Recessive Juvenile Parkinsonism. Parkin is a ubiquitin E3 ligase. Mutations in parkin are common, accounting for more than half of all cases with recessive familial PD, particularly those with very early age of onset. Although parkin null mice generated in different laboratories show no evidence of dopaminergic degeneration, there is strong experimental support for the neuroprotective role of parkin. Parkin has been shown to protect neurons from a surprisingly wide variety of insults, although the mechanisms of parkin-mediated neuroprotection remain obscure. Arrestin2 has been previously shown to bind Mdm2, another E3 ubiquitin ligase that is critical for cell survival, since it participates in the degradation of p53, a pro-apoptotic transcription factor. We found that parkin promotes Mdm2 binding to arrestin2, which may be the mechanism of the cytoprotective activity of both parkin and arrestin2. We are currently investigating the role of arrestins and arrestin-parkin interaction in cell death and survival. This work may yield important insights into the mechanism of neuronal death in Parkinson’s disease and lead to the development of neuroprotective therapies for this disorder.