Research
Research Overview
Ribosomes are large macromolecular machines that catalyze protein synthesis in all cells. Their function depends on communication between distant sites on the ribosomes, and thus requires correct assembly from the 80+ constituent components. In our lab, we study how assembly occurs within cells, how it is regulated, and what the mechanism are for quality control of assembly, and repair and quality control after assembly is complete, during translation. Importantly, when cells cannot assembly the number of ribosomes needed for normal growth, or if the ribosomes that are assembled are compromised, diseases such as Diamond Blackfan anemia, 5q- syndrome and congenital asplenia occur. These are also associated with a ~30-fold higher cancer incidence. Conversely, cancer cells accumulate misassembled ribosomes, either due to mutations in the ribosome components, or the assembly machinery. We have used these cancer-associated mutations to better understand how the assembly process is quality-controlled, and how these checkpoints are bypassed in cancer. We are very interested in studying how these “cancer-ribosomes” perturb translation.
Ribosome assembly in eukaryotic cells requires a large macromolecular machinery, comprising over 200 protein and RNA factors, which transiently associate with assembling ribosomes to facilitate the transcription of pre-rRNA, the modification and cleavage of rRNA precursors, the folding of rRNA as well as the binding of r-proteins. They also provide an interface to allow for regulation and quality control. While most of these assembly factors (AFs) are essential (~ 20% of essential genes in yeast are involved in ribosome assembly), and conserved from yeast to humans, their function remains often unknown.
We use a combination of approaches – including biochemistry, protein engineering, cryo-EM, yeast genetics and RNA and ribo-seq analysis – to study how ribosomes are assembled, what quality control mechanisms are in place to ensure only correctly and fully matured ribosomes are released into the translating pool, and how bypass of these steps, which produces misassembled ribosomes, leads to disease.
Quality Control During 40S assembly
Much of our efforts have concentrated on late cytoplasmic steps in the assembly of the small ribosomal subunit, which we have dissected in molecular detail. We have shown that assembly is coupled to quality control via a translation-like cycle (Strunk et al., Cell 2012). In this cycle the translation initiation factor eIF5B promotes joining of pre-40S subunits with 60S subunits. Maturation occurs in these 80S-like particles. However, because they contain neither mRNA or tRNA, they do not produce protein. Release of newly assembled 40S subunits into the translating pool then requires the termination factors Rli1 and Dom34 (Figure 1).
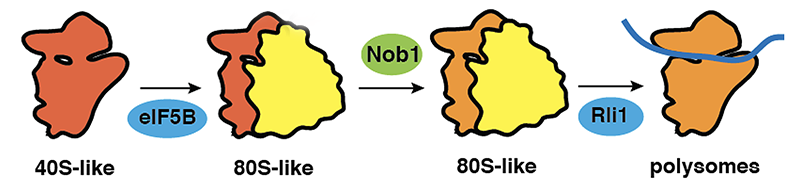
Figure 1: Testing maturing 40S ribosomes in a translation-like cycle. 40S precursors (dark orange) join 60S subunits (yellow) in a reaction catalyzed by eIF5B. The nuclease Nob1 promotes 40S maturation (to light orange 40S subunits), before Rli1 separates 40S and 60S subunits.
By studying mutants from cancer cells we have uncovered mechanisms of quality control that ensure that nascent small subunits are competent to carry out key functions of the subunit: reading frame maintenance and start codon selection. Moreover, we have also shown how kinases ensure that only fully matured subunits enter the translating pool. We are currently testing the idea that ribosome modifications are checkmarks that signify correctly assembled ribosomes and that misassembled ribosomes are a common feature of all cancer cells.
How do rRNAs fold efficiently?
We have recently shown that 3-helix junctions represent common barriers to the folding of 18S rRNA, and demonstrated how assembly factors ameliorate these problems by isolating junctions to avoid kinetic traps (Huang & Karbstein, PNAS 2021). Conceptually related, it has been shown that while early assembly steps can proceed via parallel pathways, it is also clear that some assembly routes accessible in vitro represent dead-ends, likely due to stable rRNA misfolding. We have now demonstrated a novel function for assembly factors in blocking the entrance to those dead-ended routes. We are interested in using 40S assembly as a system to better understand which rRNA sequences can fold correctly in vivo, and how assembly factors and snoRNAs can promote this process.
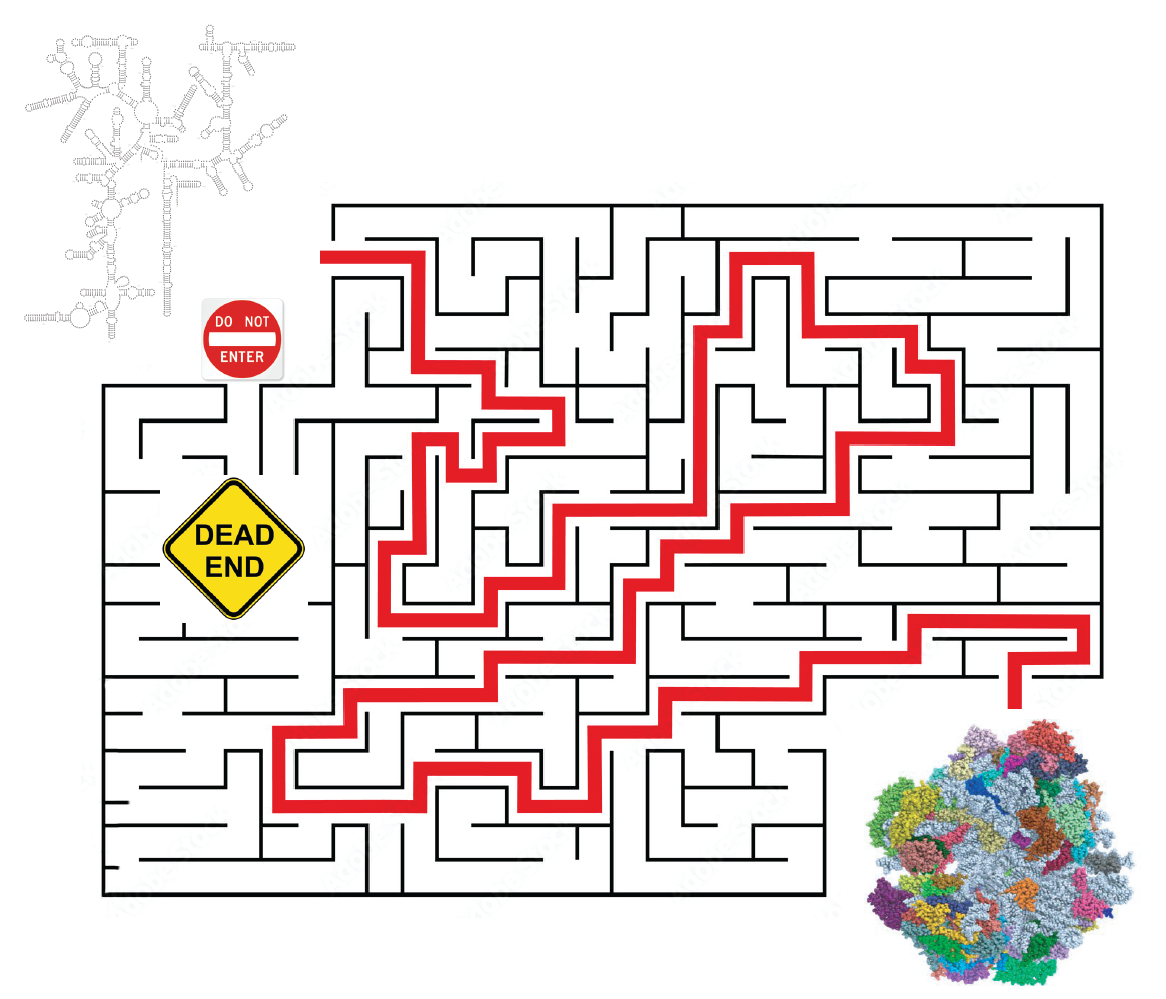
Figure 2: Assembly factors block non-productive entrances to the folding maze.
How do specialized ribosomes arise and lead to disease?
Ribosome heterogeneity has been recently documented. While not all of these examples may represent functionally relevant specialization, and could instead also reflect accidents, damage or even degradation intermediates, some examples of physiologically relevant heterogeneity are well documented (See Ferretti & Karbstein, 2019, and Yang & Karbstein, 2024 for reviews).
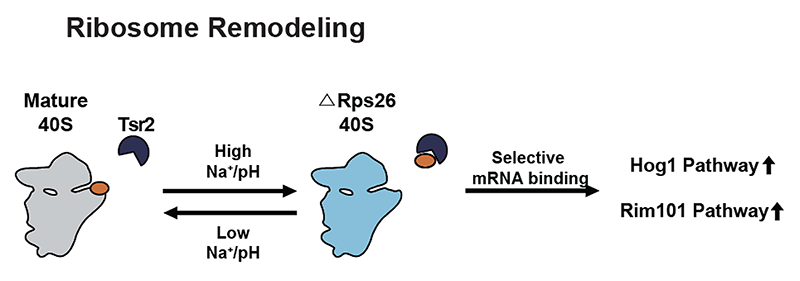
Figure 3: High intracellular Na+ concentrations or pH leads to ribosome remodeling by Tsr2-dependent dissociation of Rps26 from 40S ribosomes, producing Rps26-deficient ribosomes that display altered mRNA preference and enable translation of mRNAs from the Hog1 and Rim101 salt and pH response pathways (Ferretti et al., 2017). Removal of the stress allows for ribosome repair by allowing reincorporation of Rps26 from the Rps26•Tsr2 complex.
While their de-novo assembly would take a prohibitively long time and would also be disfavored under all stress conditions, we have recently shown that ribosomes can be rapidly and reversibly remodeled under stress by chaperone-dependent release of ribosomal proteins Rps26 (Yang&Karbstein, 2021) and Rpl10 (Yang et al., 2023), establishing a novel paradigm.
We are now studying how specifically Rps26-deficient ribosomes alter protein homeostasis to support the osmotic stress response via translational reprogramming. In addition, we and others have also demonstrated how defective ribosomes can arise in cancer cells or other diseases, either from insufficiency of assembly factors, or mutations that allow bypass of quality control checkpoints. We are interested in better understanding how these contribute to disease.
How are dysfunctional ribosomes identified during translation to be repaired or degraded?
We have recently shown that dysfunctional ribosomes are identified by the collisions they incur with faster-traveling functional ribosomes, to then be degraded (Parker et al., PLoS Biology, 2024). In addition, ribosomes with oxidatively damaged ribosomal proteins Rps26 and Rpl10 can be repaired by chaperone-mediated release of the damaged proteins and incorporation of newly-made functional proteins (Yang et al., 2023). What determines which of these routes is taken remains unclear, as are the mechanisms by which the damaged ribosomes are removed from the translating pool prior to repair.
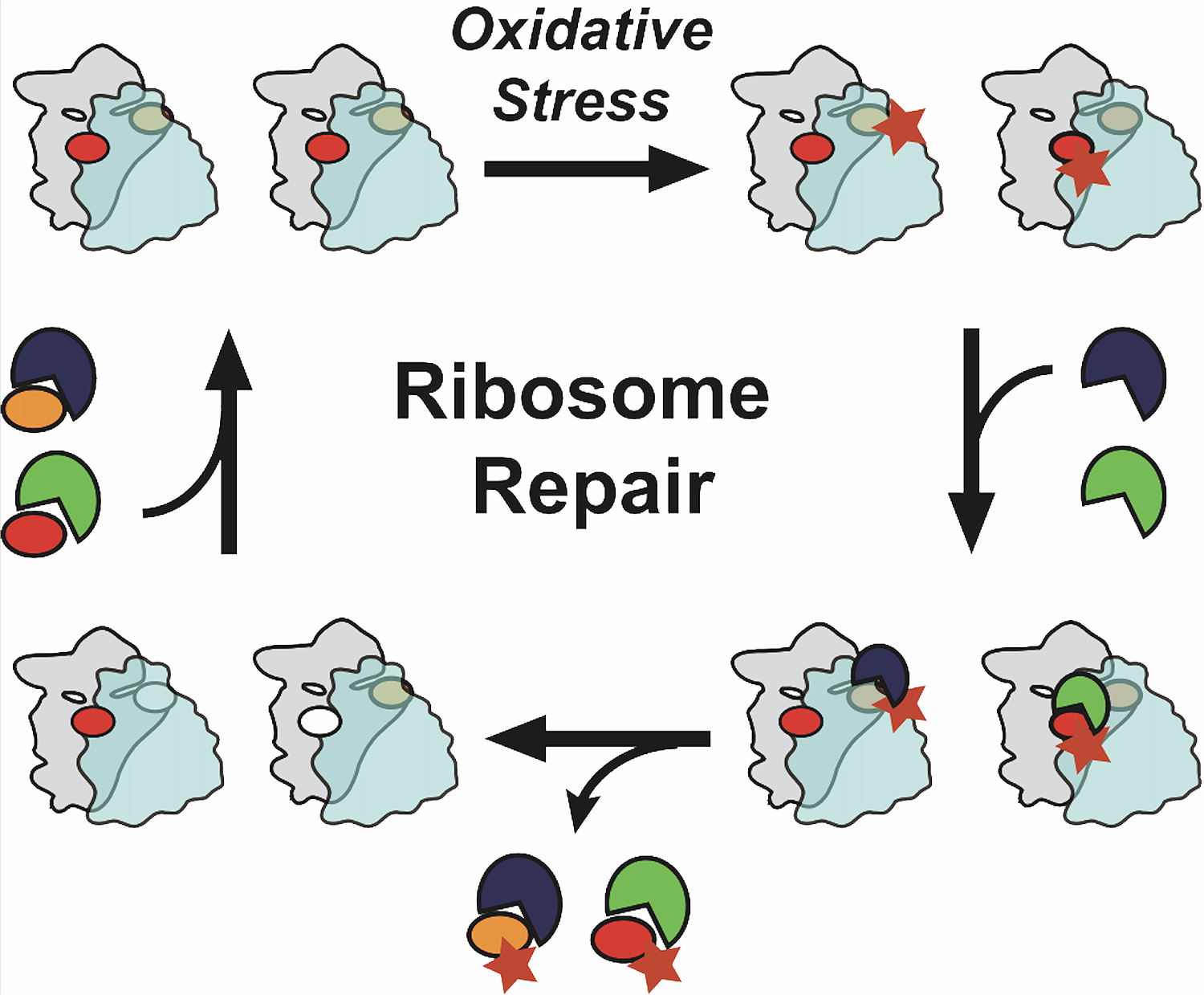
Figure 4: Oxidative stress preferentially oxidizes Rps26 and Rpl10. The damaged ribosomes are removed from the translating pool, and chaperones bind these idle 80S ribosomes to release the damaged ribosomal proteins and allow for repair with newly made ribosomal proteins.